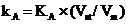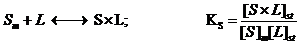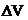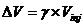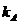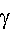Project description Scientific context and motivation.
More often, samples submitted to the chemical analysis are characterized by three main features: a) complex matrices; b); very low amounts of target
analytes to be assayed; c) availability of small sample amounts.
The extreme complexity of the matrixes is usually solved by the use of a separation technique, chromatography being frequently considered for such a goal.
Even considering the high separation power of the chromatographic processes, additional sample preparation techniques are often required to isolate analytes
from matrixes potentially affecting the success of the chromatographic separation and/or detection. Usually, analytes are isolated in organic solvents (i.e. after
liquid-liquid or solid phase extraction procedures), the compatibility of the extraction media with the mobile phase being essential for the success of the
separation.
The assay of trace amounts from target compounds is solved through the use of sensitive (and selective, as selectivity naturally enhances on sensitivity
through reduction of the background noise amplitude) detectors. In some instances, the instrumental sensitivity still may be found as inadequate, and
consequently concentration techniques are required to ensure the required sensitivity imposed thresholds. In such conditions, isolation techniques involve
concentration steps (selective removal of the isolation media until a dried residue is obtained, followed by its re-dissolution in an appropriate volume of a
diluent).
The simplest and the most straightforward way for enhancing on the sensitivity of an analytical chromatographic method is the increase of the injected
sample volume. The golden rule of the thumb for large volume injection (LVI) in liquid chromatography is that the sample diluent should be entirely miscible to
and weaker than the mobile phase composition at the beginning of the separation [1,2]. Effects of poorly controlled LVI conditions materialize in peak broadening
and/or symmetry distortions [3,4]. Although the phenomena of peak focusing after large volume injection are more often discussed in terms of the different
solubilities of the analytes in the sample diluent and the mobile phase, additional factors such as pH, composition (i.e. presence or absence of an ion pair agent)
and relative viscosities should also be considered [5-7]. Old concepts, such as elutropic character or solvent strength are still being used in practice, but they do
not offer a good insight of the complex process occurring during injection and retention processes within the chromatographic column. The role of sample solvent
strength in fast-gradient LC has been emphasized recently by Layne et al [8], demonstrating that earlier-eluting analytes are much more affected by the sample
solvent strength than the later-eluting ones.
Some practical solutions to accommodate stronger diluents to large volume injection - reversed phase liquid chromatography (RPLC) through application
of pulsed elution gradients [9] or focusing analytes in the stationary phase have been already proposed [10].
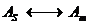
Injection of diluents stronger than the mobile phase was first pioneered by Loesser [11].
The possibility of LVI of diluents non-miscible with the mobile phase in RPLC was investigated for the first time by our team, some of the major theoretical
aspects being addressed in ref. [12]. Two points of view are usually taken into account when discussing the RP separation mechanism: the liquid-liquid partition
and the adsorption models. The first approach relies on explanation of the influence of various parameters (pH, organic modifier nature/content, salting effects)
on the retention parameters (retention time, capacity factors, peak symmetry). However, assumption that the retention takes place mainly as an adsorption
process has found agreement in many experiments, and, for instance, Guiochon and coworkers successfully developed many theoretic approaches on the topic
[13-15]. From the experimental point of view, LVI of immiscible diluents leads to a progresive reduction of the retention of the analyte A with the increase of the
injection volume [17, 18]. A linear functional dependence between the retention factor (kA) of the analyte and the injected volume (Vinj) is thus obtained. Assuming
the partion model of RP separation mechanism, the transfer of the entire volume of diluent into the hydrocarbonaceous coverage of the surface of the silicagel
based material after injection results in creation of a „new” stationary phase, with a higher carbon content, available for the analyte A. In such a case, the volume
of the stationary phase (Vst) would be increased to and consequently the fundamental relationship in LC:
(1)
would lead to a higher retention (a higher capacity factor – kA), while the equilibrium constant KA could be fairly considered constant. The stationary phase ratio
Vst/Vm would increase with the increase of the diluent volume loaded to the column. Nevertheless, the experiments revealed the oposite situation.
The adsorption mechanism starts from the equilibria involving interactions between the hydrocarbonaceus ligand, denoted L (i.e. hydrocarbonate chain
bound on the silicagel surface), the molecules of the diluent and the target analyte. The analyte (A) diffuses out of the injection plug (the diluent S) in the mobile
phase (m), according to equilibrium (2). The analyte and the diluent are competing for adsorption to the hydrocarbonaceous ligand in the stationary phase (L),
according to following processes (3) and (4):
(2)
(3)
(4)
where st, m indexes refer to the stationary and mobile phases, respectively.
Due to the high amount of S loaded in the column and its higher hydrophobic character compared to A, equilibrium (2) is shifted towards right and
equilibrium (3) toward left, as long as the entire amount of the diluent is adsorbed onto L. When the diluent has increased viscosity than the mobile phase, the
low viscosity mobile phase penetrates into the viscous plug, sending “fingers” of greater mobility ahead of the normal position of the front. As a consequence,
typical “waves” at the rear of the peaks may be observed. The process was exhaustively described in [7]. In this way, a portion from the stationary phase ( ) in
the head of column is available only to the diluent. V is proportional to Vinj, according to relation , where is a constant. Consequently, compound A
participates to the retention mechanism in the chromatographic column based on a remaining available stationary phase volume equal to (considering
the initial volume of the stationary phase as Vst). Based on these assumptions, the dependence between the retention factor of compound A ( ) and Vinj, for a
mobile phase having the volume Vm, is illustrated by the following relationship:
(5)
This relationship fully explains the experimental data observed for different analytes and diluents.
According to existing experience, succesful LVI of immiscible diluents may be possible if the following conditions are simultaneously fulfilled: a) the
retention factor of the diluent front is larger than the retention factors characterizing the target compounds; b) the solubility of the diluent in the mobile phase
should be low enough to force the saturation of the stationary phase immediately after injection; c) the diluent plug from a previous injection is eliminated from
the column before a subsequent one; d) the difference between the viscosities of the diluent and the mobile phase does not induce fingering effects potentially
producing peak distortion; e) the chromatographic resolution between target compounds should be high enough to compensate the reduction of the retention
produced through saturation of the stationary phase by the diluent. Additionally, if the diluent originated from a previous sample preparation step (i.e. liquid-
liquid extraction or desorption solvent for a SPE process), it should extract the target compounds with high yields from the original matrix or from the adsorbent.
Our findings were concretized in some applications relating to quality control of pharmaceuticals [16, 18] and bioanalysis [19]. Another application from
the field of drug development [20] has been also recently published, the importance of our first theoretical approach from publication [12] being recognized and
highlighted. Information about LVI in hydrophilic interaction liquid chromatography separation mechanism is addressed in reference [21].
To resume, the choice of out proposal for a research project having as subject LVI of immiscible diluents in LC is based on the following aspects: a) the
processes is directly related to fundamental aspects of chromatographic separation mechanisms; b) the area of interest is extremely large, as it should reflect the
mutual relationships between structural specificity of the analytes, stationary phases, mobile phases and separation mechanisms; c) a better insight of the
intimate mechanisms related to the process potentially leads to identification of immiscible solvents environmentally friendly, making an approach to the green
chemistry; d) application of LVI of immiscible diluents in analytical practice leads to a direct increase of sensitivity and naturally produces a link between usual
extraction practices used in sample preparation to the chromatographic techniques, allowing on-line automation; e) due to the straightforward characteristics, the
practice may be valuable in bioanalysis, the technique accommodating well with very large number of samples; f) last but not least, the technique was recently
introduced in the attention of the chromatographic community, and we were the first ones publishing about this topic.
Objectives
Objective 1.
Obtaining insights on the process based on LVI of immiscible solvents in liquid chromatography.
Activity 1. Checking for different diluents (eventually belonging to homologous series);
Activity 2. Checking for different mixtures of solvents used as immiscible diluents.
Activity 3. Checking for the influence of temperature on LVI of immiscible diluent process;
Activity 4. Checking for the flow rate influence on LVI of immiscible diluent process;
Activity 5. Checking for different analytes exhibiting hydrophobic character over a wide interval (large log P interval);
Activity 6. Checking for different stationary phases used under the reversed phase separation mechanism (i.e. octyl, octadecyl, amide-octadecyl, phenyl,
pentafluorophenyl chemically modified silicagels);
Activity 7. Checking for different morphologies of the packing material (i.e. 5 m particle sized materials, sub 2 m particle sized materials, fused core
materials, syntherized materials);
Activity 8. Checking for different polymeric stationary phases (i.e. SDVB, Oasis);
Activity 9. Checking for different organic components of the mobile phase (in RP mechanism) accommodating well with LVI of immiscible diluent processes;
Activity 10. Checking for different separation mechanisms (i.e. normal phase, aqueous normal phase, HILIC).
Objective 2.
Proposal of an adsorption model of the RP chromatographic mechanism, based on multiple layer formation (Freundlich isotherm).
Activity 1. Study of the elution shape of the diluent front after an LVI process by means of RID detection.
Objective 3.
The possibility of characterization of the hydrophobic characteristics of organic compounds based on their ability to perform LVI in
different immiscible diluents, without peak broadening and/or distortion effects.
Activity 1. Identification of series of immiscible diluents generating log P progressive scales;
Activity 2. Identification of the general LVI conditions allowing the direct comparison between the hydrophobic character of the analytes and the diluents;
Activity 3. Correlation between the hydrophobic characteristics of a set of compounds determined from the LVI of immiscible diluents procedure to
experimentally determined or computed log P values.
Objective 4.
Identification of “green” solvents to be used during usual sample preparation steps (i.e. LLE or SPE) which can further play the role of
immiscible diluents for LVI in LC separations.
Objective 5.
Identification of on-line automation issues for accommodation of the extractive sample preparation techniques to LVI of immiscible
diluents in LC
Activity 1. automation of LLE to LVI of immiscible diluents in LC;
Activity 2. automation of SPE to LVI of immiscible diluents in LC;
Objective 6.
Identification of potential application fields for LVI of immiscible solvents:
Activity 1. applications in bioanalysis;
Activity 2. applications in the quality control of pharmaceuticals;
Activity 3. applications in environmental quality control;
Activity 4. applications in food analysis.
To resume the above mentioned objectives, two principal directions should be highlighted: a) the fundamental theoretical aspects of the LVI technique; b)
the applicative aspects relating to LVI (indissolubly related to sensitivity increase and support of the straightforward character linking sample preparation steps to
the chromatographic analysis, resulting in a high throughput of analytical approaches). The proposed topic is important because it refers to new aspects revealed
recently in literature (most of them belong to the research team proposing the project). Importance of the proposed subject should be related not only to the gain
of the basic knowledge relating to the chromatographic separation processes, but also with evident experimental resulting advantages (increased sensitivity,
automation capabilities, high throughput). Considering the novelty of the proposed topic as well as the integrated theoretical/applicative character, dissemination
(by means of publication in highly ranked journals, as well as participation to international symposia) of the results should be considered as a permanent
objective, denoted as Objective 7. As the successful development of the project directly rely on the existence of a corresponding web site, it creation and upgrading
will be referred as Objective 8.
References
1.
Snyder L.R., Kirkland J.J. Introduction to modern liquid chromatography. 2nd ed. Wiley, New York, (1979).
2.
Snyder L.R., Kirkland J.J., Glajch J.L., Practical HPLC method development. Wiley, New York, (1988).
3.
Dolan J.W. Broad peaks, LC-GC North Am., 22, 26-30 (2004).
4.
Pedersen K., Dolan J.W., A picture is worth a thousand Words, LC-GC Europe, 24(2), 72-76 (2011).
5.
Keunchkarian S., Reta M., Romero L., Castells C.B. Effect of sample solvent on the chromatographic peak shape of analytes eluted under reversed-phase
liquid chromatographic conditions, J. Chromatogr. A, 1119(1/2), 20-28 (2006).
6.
Castells C.B., Castells R.C. Peak distortion in reversed-phase liquid chromatography as a consequence of viscosity differences between sample solvent and
mobile phase, J. Chromatogr. A, 805(1/2), 55-61 (1998).
7.
Cherrak D., Guernet E., Cardot P., Herrenknecht C., Czok M., Viscous fingering: a systematic study of viscosity effects in methanol-isopropanol systems,
Chromatographia, 46, 647-654 (1997).
8.
Layne J., Farcas T., Rustamov I., Ahmed F., Volume load capacity in fast gradient liquid chromatography: effect of sample solvent composition and
injection volume on chromatographic performance, J. Chromatogr. A, 913, 233-242 (2001).
9.
Li Y., Suna Y., Dua F., Yuanc K., Li C., Pulse gradient, large-volume injection, high throughput ultra-performance liquid chromatographic/tandem mass
spectrometry bioanalysis for measurement of plasma amrubicin and its metabolite amrubicinol, J. Chromatogr. A, 1193, 109-116 (2008).
10.
Lovin I., Albu F., Tache F., David V., Medvedovici A. Solvent and salting effects on sample preparation for the determination of fenofibric acid in human
plasma by HPLC-DAD, Microchem. J. 75, 179-187 (2003).
11.
Loeser E, Drumm P. Using strong injection solvents with 100% aqueous mobile phase in RP-LC, J. Sep. Sci. 29(18), 2847-2852 (2006).
12.
Medvedovici A., David Vasile, David Victor, Georgita C. Retention phenomena induced by large volume injection of solvents non-miscible with the mobile
phase in reversed phase liquid chromatography, J. Liq. Chromatogr. Relat. Technol. 30, 199-213 (2007).
13.
Gritti F., Piatkowski W., Guiochon G. Study of the mass transfer kinetics in a monolithic column, J. Chromatogr. A, 983, 51-71 (2003).
14.
Miyabe K., Guiochon G. New model of surface diffusion in reversed phase liquid chromatography, J. Chromatogr. A, 961, 23 – 33 (2002).
15.
Ahmad T., Gritti F., Lin B., Guiochon G., Single component shock layer analysis in elution chromatography, Anal. Chem., 76, 977 – 984 (2004).
16.
David V., Barcutean C., Georgita C., Medvedovici A. Non-miscible solvent large volume injection – HPLC/DAD method for determination of butylated
hydroxyanisole in lovastatin and simvastatin pharmaceutical formulations, Rev. Roum. Chim., 5, 445-451 (2006) .
17.
Udrescu S., Medvedovici A., David V., Effect of large volume injection of hydrophobic solvents on the retention of less hydrophobic pharmaceutical solutes
in RP-LC, J. Sep. Sci., 31, 2939-2945 (2008).
18.
Udrescu S., Sora I.D., David V., Medvedovici A., Large volume injection of hexane solutions in RPLC/UV to enhance on sensitivity of the assay of ginkgolic
acids in Ginkgo Biloba standardized extracts, J. Liq. Chromatogr. Relat. Technol., 33,133-149 (2010).
19.
Udrescu S., Sora I.D., Albu F., David V., Medvedovici A. Large volume injection of 1-octanol as sample diluent in reversed phase liquid chromatography:
Application in bioanalysis for assaying of indapamide in whole blood, J. Pharm. Biomed. Anal., 54(5), 1163–1172 (2011).
20.
Loeser E., Babiak S., Drumm P., Water-immiscible solvents as diluents in reversed-phase liquid chromatography, J. Chromatogr. A, 1216, 3409-3412
(2009).
21.
Ruta J., Rudaz S., McCalley D.V., Veuthey J.L., Guillaume D. A systemathic investigation of the effect of sample diluent on peak shape in hydrophilic
interaction liquid chromatography, J. Chromatogr. A, 1217, 8230-8240 (2010).
Scientific context and motivation.
More often, samples submitted to the chemical analysis are characterized by three main features: a) complex matrices; b); very low amounts of target
analytes to be assayed; c) availability of small sample amounts.
The extreme complexity of the matrixes is usually solved by the use of a separation technique, chromatography being frequently considered for such a goal.
Even considering the high separation power of the chromatographic processes, additional sample preparation techniques are often required to isolate analytes
from matrixes potentially affecting the success of the chromatographic separation and/or detection. Usually, analytes are isolated in organic solvents (i.e. after
liquid-liquid or solid phase extraction procedures), the compatibility of the extraction media with the mobile phase being essential for the success of the
separation.
The assay of trace amounts from target compounds is solved through the use of sensitive (and selective, as selectivity naturally enhances on sensitivity
through reduction of the background noise amplitude) detectors. In some instances, the instrumental sensitivity still may be found as inadequate, and
consequently concentration techniques are required to ensure the required sensitivity imposed thresholds. In such conditions, isolation techniques involve
concentration steps (selective removal of the isolation media until a dried residue is obtained, followed by its re-dissolution in an appropriate volume of a
diluent).
The simplest and the most straightforward way for enhancing on the sensitivity of an analytical chromatographic method is the increase of the injected
sample volume. The golden rule of the thumb for large volume injection (LVI) in liquid chromatography is that the sample diluent should be entirely miscible to
and weaker than the mobile phase composition at the beginning of the separation [1,2]. Effects of poorly controlled LVI conditions materialize in peak broadening
and/or symmetry distortions [3,4]. Although the phenomena of peak focusing after large volume injection are more often discussed in terms of the different
solubilities of the analytes in the sample diluent and the mobile phase, additional factors such as pH, composition (i.e. presence or absence of an ion pair agent)
and relative viscosities should also be considered [5-7]. Old concepts, such as elutropic character or solvent strength are still being used in practice, but they do
not offer a good insight of the complex process occurring during injection and retention processes within the chromatographic column. The role of sample solvent
strength in fast-gradient LC has been emphasized recently by Layne et al [8], demonstrating that earlier-eluting analytes are much more affected by the sample
solvent strength than the later-eluting ones.
Some practical solutions to accommodate stronger diluents to large volume injection - reversed phase liquid chromatography (RPLC) through application
of pulsed elution gradients [9] or focusing analytes in the stationary phase have been already proposed [10].
Injection of diluents stronger than the mobile phase was first pioneered by Loesser [11].
The possibility of LVI of diluents non-miscible with the mobile phase in RPLC was investigated for the first time by our team, some of the major theoretical
aspects being addressed in ref. [12]. Two points of view are usually taken into account when discussing the RP separation mechanism: the liquid-liquid partition
and the adsorption models. The first approach relies on explanation of the influence of various parameters (pH, organic modifier nature/content, salting effects)
on the retention parameters (retention time, capacity factors, peak symmetry). However, assumption that the retention takes place mainly as an adsorption
process has found agreement in many experiments, and, for instance, Guiochon and coworkers successfully developed many theoretic approaches on the topic
[13-15]. From the experimental point of view, LVI of immiscible diluents leads to a progresive reduction of the retention of the analyte A with the increase of the
injection volume [17, 18]. A linear functional dependence between the retention factor (kA) of the analyte and the injected volume (Vinj) is thus obtained. Assuming
the partion model of RP separation mechanism, the transfer of the entire volume of diluent into the hydrocarbonaceous coverage of the surface of the silicagel
based material after injection results in creation of a „new” stationary phase, with a higher carbon content, available for the analyte A. In such a case, the volume
of the stationary phase (Vst) would be increased to and consequently the fundamental relationship in LC:
(1)
would lead to a higher retention (a higher capacity factor – kA), while the equilibrium constant KA could be fairly considered constant. The stationary phase ratio
Vst/Vm would increase with the increase of the diluent volume loaded to the column. Nevertheless, the experiments revealed the oposite situation.
The adsorption mechanism starts from the equilibria involving interactions between the hydrocarbonaceus ligand, denoted L (i.e. hydrocarbonate chain
bound on the silicagel surface), the molecules of the diluent and the target analyte. The analyte (A) diffuses out of the injection plug (the diluent S) in the mobile
phase (m), according to equilibrium (2). The analyte and the diluent are competing for adsorption to the hydrocarbonaceous ligand in the stationary phase (L),
according to following processes (3) and (4):
(2)
(3)
(4)
where st, m indexes refer to the stationary and mobile phases, respectively.
Due to the high amount of S loaded in the column and its higher hydrophobic character compared to A, equilibrium (2) is shifted towards right and
equilibrium (3) toward left, as long as the entire amount of the diluent is adsorbed onto L. When the diluent has increased viscosity than the mobile phase, the
low viscosity mobile phase penetrates into the viscous plug, sending “fingers” of greater mobility ahead of the normal position of the front. As a consequence,
typical “waves” at the rear of the peaks may be observed. The process was exhaustively described in [7]. In this way, a portion from the stationary phase ( ) in
the head of column is available only to the diluent. V is proportional to Vinj, according to relation , where is a constant. Consequently, compound A
participates to the retention mechanism in the chromatographic column based on a remaining available stationary phase volume equal to (considering
the initial volume of the stationary phase as Vst). Based on these assumptions, the dependence between the retention factor of compound A ( ) and Vinj, for a
mobile phase having the volume Vm, is illustrated by the following relationship:
(5)
This relationship fully explains the experimental data observed for different analytes and diluents.
According to existing experience, succesful LVI of immiscible diluents may be possible if the following conditions are simultaneously fulfilled: a) the
retention factor of the diluent front is larger than the retention factors characterizing the target compounds; b) the solubility of the diluent in the mobile phase
should be low enough to force the saturation of the stationary phase immediately after injection; c) the diluent plug from a previous injection is eliminated from
the column before a subsequent one; d) the difference between the viscosities of the diluent and the mobile phase does not induce fingering effects potentially
producing peak distortion; e) the chromatographic resolution between target compounds should be high enough to compensate the reduction of the retention
produced through saturation of the stationary phase by the diluent. Additionally, if the diluent originated from a previous sample preparation step (i.e. liquid-
liquid extraction or desorption solvent for a SPE process), it should extract the target compounds with high yields from the original matrix or from the adsorbent.
Our findings were concretized in some applications relating to quality control of pharmaceuticals [16, 18] and bioanalysis [19]. Another application from
the field of drug development [20] has been also recently published, the importance of our first theoretical approach from publication [12] being recognized and
highlighted. Information about LVI in hydrophilic interaction liquid chromatography separation mechanism is addressed in reference [21].
To resume, the choice of out proposal for a research project having as subject LVI of immiscible diluents in LC is based on the following aspects: a) the
processes is directly related to fundamental aspects of chromatographic separation mechanisms; b) the area of interest is extremely large, as it should reflect the
mutual relationships between structural specificity of the analytes, stationary phases, mobile phases and separation mechanisms; c) a better insight of the
intimate mechanisms related to the process potentially leads to identification of immiscible solvents environmentally friendly, making an approach to the green
chemistry; d) application of LVI of immiscible diluents in analytical practice leads to a direct increase of sensitivity and naturally produces a link between usual
extraction practices used in sample preparation to the chromatographic techniques, allowing on-line automation; e) due to the straightforward characteristics, the
practice may be valuable in bioanalysis, the technique accommodating well with very large number of samples; f) last but not least, the technique was recently
introduced in the attention of the chromatographic community, and we were the first ones publishing about this topic.
Objectives
Objective 1.
Obtaining insights on the process based on LVI of immiscible solvents in liquid chromatography.
Activity 1. Checking for different diluents (eventually belonging to homologous series);
Activity 2. Checking for different mixtures of solvents used as immiscible diluents.
Activity 3. Checking for the influence of temperature on LVI of immiscible diluent process;
Activity 4. Checking for the flow rate influence on LVI of immiscible diluent process;
Activity 5. Checking for different analytes exhibiting hydrophobic character over a wide interval (large log P interval);
Activity 6. Checking for different stationary phases used under the reversed phase separation mechanism (i.e. octyl, octadecyl, amide-octadecyl, phenyl,
pentafluorophenyl chemically modified silicagels);
Activity 7. Checking for different morphologies of the packing material (i.e. 5 m particle sized materials, sub 2 m particle sized materials, fused core
materials, syntherized materials);
Activity 8. Checking for different polymeric stationary phases (i.e. SDVB, Oasis);
Activity 9. Checking for different organic components of the mobile phase (in RP mechanism) accommodating well with LVI of immiscible diluent processes;
Activity 10. Checking for different separation mechanisms (i.e. normal phase, aqueous normal phase, HILIC).
Objective 2.
Proposal of an adsorption model of the RP chromatographic mechanism, based on multiple layer formation (Freundlich isotherm).
Activity 1. Study of the elution shape of the diluent front after an LVI process by means of RID detection.
Objective 3.
The possibility of characterization of the hydrophobic characteristics of organic compounds based on their ability to perform LVI in
different immiscible diluents, without peak broadening and/or distortion effects.
Activity 1. Identification of series of immiscible diluents generating log P progressive scales;
Activity 2. Identification of the general LVI conditions allowing the direct comparison between the hydrophobic character of the analytes and the diluents;
Activity 3. Correlation between the hydrophobic characteristics of a set of compounds determined from the LVI of immiscible diluents procedure to
experimentally determined or computed log P values.
Objective 4.
Identification of “green” solvents to be used during usual sample preparation steps (i.e. LLE or SPE) which can further play the role of
immiscible diluents for LVI in LC separations.
Objective 5.
Identification of on-line automation issues for accommodation of the extractive sample preparation techniques to LVI of immiscible
diluents in LC
Activity 1. automation of LLE to LVI of immiscible diluents in LC;
Activity 2. automation of SPE to LVI of immiscible diluents in LC;
Objective 6.
Identification of potential application fields for LVI of immiscible solvents:
Activity 1. applications in bioanalysis;
Activity 2. applications in the quality control of pharmaceuticals;
Activity 3. applications in environmental quality control;
Activity 4. applications in food analysis.
To resume the above mentioned objectives, two principal directions should be highlighted: a) the fundamental theoretical aspects of the LVI technique; b)
the applicative aspects relating to LVI (indissolubly related to sensitivity increase and support of the straightforward character linking sample preparation steps to
the chromatographic analysis, resulting in a high throughput of analytical approaches). The proposed topic is important because it refers to new aspects revealed
recently in literature (most of them belong to the research team proposing the project). Importance of the proposed subject should be related not only to the gain
of the basic knowledge relating to the chromatographic separation processes, but also with evident experimental resulting advantages (increased sensitivity,
automation capabilities, high throughput). Considering the novelty of the proposed topic as well as the integrated theoretical/applicative character, dissemination
(by means of publication in highly ranked journals, as well as participation to international symposia) of the results should be considered as a permanent
objective, denoted as Objective 7. As the successful development of the project directly rely on the existence of a corresponding web site, it creation and upgrading
will be referred as Objective 8.
References
1.
Snyder L.R., Kirkland J.J. Introduction to modern liquid chromatography. 2nd ed. Wiley, New York, (1979).
2.
Snyder L.R., Kirkland J.J., Glajch J.L., Practical HPLC method development. Wiley, New York, (1988).
3.
Dolan J.W. Broad peaks, LC-GC North Am., 22, 26-30 (2004).
4.
Pedersen K., Dolan J.W., A picture is worth a thousand Words, LC-GC Europe, 24(2), 72-76 (2011).
5.
Keunchkarian S., Reta M., Romero L., Castells C.B. Effect of sample solvent on the chromatographic peak shape of analytes eluted under reversed-phase
liquid chromatographic conditions, J. Chromatogr. A, 1119(1/2), 20-28 (2006).
6.
Castells C.B., Castells R.C. Peak distortion in reversed-phase liquid chromatography as a consequence of viscosity differences between sample solvent and
mobile phase, J. Chromatogr. A, 805(1/2), 55-61 (1998).
7.
Cherrak D., Guernet E., Cardot P., Herrenknecht C., Czok M., Viscous fingering: a systematic study of viscosity effects in methanol-isopropanol systems,
Chromatographia, 46, 647-654 (1997).
8.
Layne J., Farcas T., Rustamov I., Ahmed F., Volume load capacity in fast gradient liquid chromatography: effect of sample solvent composition and
injection volume on chromatographic performance, J. Chromatogr. A, 913, 233-242 (2001).
9.
Li Y., Suna Y., Dua F., Yuanc K., Li C., Pulse gradient, large-volume injection, high throughput ultra-performance liquid chromatographic/tandem mass
spectrometry bioanalysis for measurement of plasma amrubicin and its metabolite amrubicinol, J. Chromatogr. A, 1193, 109-116 (2008).
10.
Lovin I., Albu F., Tache F., David V., Medvedovici A. Solvent and salting effects on sample preparation for the determination of fenofibric acid in human
plasma by HPLC-DAD, Microchem. J. 75, 179-187 (2003).
11.
Loeser E, Drumm P. Using strong injection solvents with 100% aqueous mobile phase in RP-LC, J. Sep. Sci. 29(18), 2847-2852 (2006).
12.
Medvedovici A., David Vasile, David Victor, Georgita C. Retention phenomena induced by large volume injection of solvents non-miscible with the mobile
phase in reversed phase liquid chromatography, J. Liq. Chromatogr. Relat. Technol. 30, 199-213 (2007).
13.
Gritti F., Piatkowski W., Guiochon G. Study of the mass transfer kinetics in a monolithic column, J. Chromatogr. A, 983, 51-71 (2003).
14.
Miyabe K., Guiochon G. New model of surface diffusion in reversed phase liquid chromatography, J. Chromatogr. A, 961, 23 – 33 (2002).
15.
Ahmad T., Gritti F., Lin B., Guiochon G., Single component shock layer analysis in elution chromatography, Anal. Chem., 76, 977 – 984 (2004).
16.
David V., Barcutean C., Georgita C., Medvedovici A. Non-miscible solvent large volume injection – HPLC/DAD method for determination of butylated
hydroxyanisole in lovastatin and simvastatin pharmaceutical formulations, Rev. Roum. Chim., 5, 445-451 (2006) .
17.
Udrescu S., Medvedovici A., David V., Effect of large volume injection of hydrophobic solvents on the retention of less hydrophobic pharmaceutical solutes
in RP-LC, J. Sep. Sci., 31, 2939-2945 (2008).
18.
Udrescu S., Sora I.D., David V., Medvedovici A., Large volume injection of hexane solutions in RPLC/UV to enhance on sensitivity of the assay of ginkgolic
acids in Ginkgo Biloba standardized extracts, J. Liq. Chromatogr. Relat. Technol., 33,133-149 (2010).
19.
Udrescu S., Sora I.D., Albu F., David V., Medvedovici A. Large volume injection of 1-octanol as sample diluent in reversed phase liquid chromatography:
Application in bioanalysis for assaying of indapamide in whole blood, J. Pharm. Biomed. Anal., 54(5), 1163–1172 (2011).
20.
Loeser E., Babiak S., Drumm P., Water-immiscible solvents as diluents in reversed-phase liquid chromatography, J. Chromatogr. A, 1216, 3409-3412
(2009).
21.
Ruta J., Rudaz S., McCalley D.V., Veuthey J.L., Guillaume D. A systemathic investigation of the effect of sample diluent on peak shape in hydrophilic
interaction liquid chromatography, J. Chromatogr. A, 1217, 8230-8240 (2010).
Scientific context and motivation.
More often, samples submitted to the chemical analysis are characterized by three main features: a) complex matrices; b); very low amounts of target
analytes to be assayed; c) availability of small sample amounts.
The extreme complexity of the matrixes is usually solved by the use of a separation technique, chromatography being frequently considered for such a goal.
Even considering the high separation power of the chromatographic processes, additional sample preparation techniques are often required to isolate analytes
from matrixes potentially affecting the success of the chromatographic separation and/or detection. Usually, analytes are isolated in organic solvents (i.e. after
liquid-liquid or solid phase extraction procedures), the compatibility of the extraction media with the mobile phase being essential for the success of the
separation.
The assay of trace amounts from target compounds is solved through the use of sensitive (and selective, as selectivity naturally enhances on sensitivity
through reduction of the background noise amplitude) detectors. In some instances, the instrumental sensitivity still may be found as inadequate, and
consequently concentration techniques are required to ensure the required sensitivity imposed thresholds. In such conditions, isolation techniques involve
concentration steps (selective removal of the isolation media until a dried residue is obtained, followed by its re-dissolution in an appropriate volume of a
diluent).
The simplest and the most straightforward way for enhancing on the sensitivity of an analytical chromatographic method is the increase of the injected
sample volume. The golden rule of the thumb for large volume injection (LVI) in liquid chromatography is that the sample diluent should be entirely miscible to
and weaker than the mobile phase composition at the beginning of the separation [1,2]. Effects of poorly controlled LVI conditions materialize in peak broadening
and/or symmetry distortions [3,4]. Although the phenomena of peak focusing after large volume injection are more often discussed in terms of the different
solubilities of the analytes in the sample diluent and the mobile phase, additional factors such as pH, composition (i.e. presence or absence of an ion pair agent)
and relative viscosities should also be considered [5-7]. Old concepts, such as elutropic character or solvent strength are still being used in practice, but they do
not offer a good insight of the complex process occurring during injection and retention processes within the chromatographic column. The role of sample solvent
strength in fast-gradient LC has been emphasized recently by Layne et al [8], demonstrating that earlier-eluting analytes are much more affected by the sample
solvent strength than the later-eluting ones.
Some practical solutions to accommodate stronger diluents to large volume injection - reversed phase liquid chromatography (RPLC) through application
of pulsed elution gradients [9] or focusing analytes in the stationary phase have been already proposed [10].
Injection of diluents stronger than the mobile phase was first pioneered by Loesser [11].
The possibility of LVI of diluents non-miscible with the mobile phase in RPLC was investigated for the first time by our team, some of the major theoretical
aspects being addressed in ref. [12]. Two points of view are usually taken into account when discussing the RP separation mechanism: the liquid-liquid partition
and the adsorption models. The first approach relies on explanation of the influence of various parameters (pH, organic modifier nature/content, salting effects)
on the retention parameters (retention time, capacity factors, peak symmetry). However, assumption that the retention takes place mainly as an adsorption
process has found agreement in many experiments, and, for instance, Guiochon and coworkers successfully developed many theoretic approaches on the topic
[13-15]. From the experimental point of view, LVI of immiscible diluents leads to a progresive reduction of the retention of the analyte A with the increase of the
injection volume [17, 18]. A linear functional dependence between the retention factor (kA) of the analyte and the injected volume (Vinj) is thus obtained. Assuming
the partion model of RP separation mechanism, the transfer of the entire volume of diluent into the hydrocarbonaceous coverage of the surface of the silicagel
based material after injection results in creation of a „new” stationary phase, with a higher carbon content, available for the analyte A. In such a case, the volume
of the stationary phase (Vst) would be increased to and consequently the fundamental relationship in LC:
(1)
would lead to a higher retention (a higher capacity factor – kA), while the equilibrium constant KA could be fairly considered constant. The stationary phase ratio
Vst/Vm would increase with the increase of the diluent volume loaded to the column. Nevertheless, the experiments revealed the oposite situation.
The adsorption mechanism starts from the equilibria involving interactions between the hydrocarbonaceus ligand, denoted L (i.e. hydrocarbonate chain
bound on the silicagel surface), the molecules of the diluent and the target analyte. The analyte (A) diffuses out of the injection plug (the diluent S) in the mobile
phase (m), according to equilibrium (2). The analyte and the diluent are competing for adsorption to the hydrocarbonaceous ligand in the stationary phase (L),
according to following processes (3) and (4):
(2)
(3)
(4)
where st, m indexes refer to the stationary and mobile phases, respectively.
Due to the high amount of S loaded in the column and its higher hydrophobic character compared to A, equilibrium (2) is shifted towards right and
equilibrium (3) toward left, as long as the entire amount of the diluent is adsorbed onto L. When the diluent has increased viscosity than the mobile phase, the
low viscosity mobile phase penetrates into the viscous plug, sending “fingers” of greater mobility ahead of the normal position of the front. As a consequence,
typical “waves” at the rear of the peaks may be observed. The process was exhaustively described in [7]. In this way, a portion from the stationary phase ( ) in
the head of column is available only to the diluent. V is proportional to Vinj, according to relation , where is a constant. Consequently, compound A
participates to the retention mechanism in the chromatographic column based on a remaining available stationary phase volume equal to (considering
the initial volume of the stationary phase as Vst). Based on these assumptions, the dependence between the retention factor of compound A ( ) and Vinj, for a
mobile phase having the volume Vm, is illustrated by the following relationship:
(5)
This relationship fully explains the experimental data observed for different analytes and diluents.
According to existing experience, succesful LVI of immiscible diluents may be possible if the following conditions are simultaneously fulfilled: a) the
retention factor of the diluent front is larger than the retention factors characterizing the target compounds; b) the solubility of the diluent in the mobile phase
should be low enough to force the saturation of the stationary phase immediately after injection; c) the diluent plug from a previous injection is eliminated from
the column before a subsequent one; d) the difference between the viscosities of the diluent and the mobile phase does not induce fingering effects potentially
producing peak distortion; e) the chromatographic resolution between target compounds should be high enough to compensate the reduction of the retention
produced through saturation of the stationary phase by the diluent. Additionally, if the diluent originated from a previous sample preparation step (i.e. liquid-
liquid extraction or desorption solvent for a SPE process), it should extract the target compounds with high yields from the original matrix or from the adsorbent.
Our findings were concretized in some applications relating to quality control of pharmaceuticals [16, 18] and bioanalysis [19]. Another application from
the field of drug development [20] has been also recently published, the importance of our first theoretical approach from publication [12] being recognized and
highlighted. Information about LVI in hydrophilic interaction liquid chromatography separation mechanism is addressed in reference [21].
To resume, the choice of out proposal for a research project having as subject LVI of immiscible diluents in LC is based on the following aspects: a) the
processes is directly related to fundamental aspects of chromatographic separation mechanisms; b) the area of interest is extremely large, as it should reflect the
mutual relationships between structural specificity of the analytes, stationary phases, mobile phases and separation mechanisms; c) a better insight of the
intimate mechanisms related to the process potentially leads to identification of immiscible solvents environmentally friendly, making an approach to the green
chemistry; d) application of LVI of immiscible diluents in analytical practice leads to a direct increase of sensitivity and naturally produces a link between usual
extraction practices used in sample preparation to the chromatographic techniques, allowing on-line automation; e) due to the straightforward characteristics, the
practice may be valuable in bioanalysis, the technique accommodating well with very large number of samples; f) last but not least, the technique was recently
introduced in the attention of the chromatographic community, and we were the first ones publishing about this topic.
Objectives
Objective 1.
Obtaining insights on the process based on LVI of immiscible solvents in liquid chromatography.
Activity 1. Checking for different diluents (eventually belonging to homologous series);
Activity 2. Checking for different mixtures of solvents used as immiscible diluents.
Activity 3. Checking for the influence of temperature on LVI of immiscible diluent process;
Activity 4. Checking for the flow rate influence on LVI of immiscible diluent process;
Activity 5. Checking for different analytes exhibiting hydrophobic character over a wide interval (large log P interval);
Activity 6. Checking for different stationary phases used under the reversed phase separation mechanism (i.e. octyl, octadecyl, amide-octadecyl, phenyl,
pentafluorophenyl chemically modified silicagels);
Activity 7. Checking for different morphologies of the packing material (i.e. 5 m particle sized materials, sub 2 m particle sized materials, fused core
materials, syntherized materials);
Activity 8. Checking for different polymeric stationary phases (i.e. SDVB, Oasis);
Activity 9. Checking for different organic components of the mobile phase (in RP mechanism) accommodating well with LVI of immiscible diluent processes;
Activity 10. Checking for different separation mechanisms (i.e. normal phase, aqueous normal phase, HILIC).
Objective 2.
Proposal of an adsorption model of the RP chromatographic mechanism, based on multiple layer formation (Freundlich isotherm).
Activity 1. Study of the elution shape of the diluent front after an LVI process by means of RID detection.
Objective 3.
The possibility of characterization of the hydrophobic characteristics of organic compounds based on their ability to perform LVI in
different immiscible diluents, without peak broadening and/or distortion effects.
Activity 1. Identification of series of immiscible diluents generating log P progressive scales;
Activity 2. Identification of the general LVI conditions allowing the direct comparison between the hydrophobic character of the analytes and the diluents;
Activity 3. Correlation between the hydrophobic characteristics of a set of compounds determined from the LVI of immiscible diluents procedure to
experimentally determined or computed log P values.
Objective 4.
Identification of “green” solvents to be used during usual sample preparation steps (i.e. LLE or SPE) which can further play the role of
immiscible diluents for LVI in LC separations.
Objective 5.
Identification of on-line automation issues for accommodation of the extractive sample preparation techniques to LVI of immiscible
diluents in LC
Activity 1. automation of LLE to LVI of immiscible diluents in LC;
Activity 2. automation of SPE to LVI of immiscible diluents in LC;
Objective 6.
Identification of potential application fields for LVI of immiscible solvents:
Activity 1. applications in bioanalysis;
Activity 2. applications in the quality control of pharmaceuticals;
Activity 3. applications in environmental quality control;
Activity 4. applications in food analysis.
To resume the above mentioned objectives, two principal directions should be highlighted: a) the fundamental theoretical aspects of the LVI technique; b)
the applicative aspects relating to LVI (indissolubly related to sensitivity increase and support of the straightforward character linking sample preparation steps to
the chromatographic analysis, resulting in a high throughput of analytical approaches). The proposed topic is important because it refers to new aspects revealed
recently in literature (most of them belong to the research team proposing the project). Importance of the proposed subject should be related not only to the gain
of the basic knowledge relating to the chromatographic separation processes, but also with evident experimental resulting advantages (increased sensitivity,
automation capabilities, high throughput). Considering the novelty of the proposed topic as well as the integrated theoretical/applicative character, dissemination
(by means of publication in highly ranked journals, as well as participation to international symposia) of the results should be considered as a permanent
objective, denoted as Objective 7. As the successful development of the project directly rely on the existence of a corresponding web site, it creation and upgrading
will be referred as Objective 8.
References
1.
Snyder L.R., Kirkland J.J. Introduction to modern liquid chromatography. 2nd ed. Wiley, New York, (1979).
2.
Snyder L.R., Kirkland J.J., Glajch J.L., Practical HPLC method development. Wiley, New York, (1988).
3.
Dolan J.W. Broad peaks, LC-GC North Am., 22, 26-30 (2004).
4.
Pedersen K., Dolan J.W., A picture is worth a thousand Words, LC-GC Europe, 24(2), 72-76 (2011).
5.
Keunchkarian S., Reta M., Romero L., Castells C.B. Effect of sample solvent on the chromatographic peak shape of analytes eluted under reversed-phase
liquid chromatographic conditions, J. Chromatogr. A, 1119(1/2), 20-28 (2006).
6.
Castells C.B., Castells R.C. Peak distortion in reversed-phase liquid chromatography as a consequence of viscosity differences between sample solvent and
mobile phase, J. Chromatogr. A, 805(1/2), 55-61 (1998).
7.
Cherrak D., Guernet E., Cardot P., Herrenknecht C., Czok M., Viscous fingering: a systematic study of viscosity effects in methanol-isopropanol systems,
Chromatographia, 46, 647-654 (1997).
8.
Layne J., Farcas T., Rustamov I., Ahmed F., Volume load capacity in fast gradient liquid chromatography: effect of sample solvent composition and
injection volume on chromatographic performance, J. Chromatogr. A, 913, 233-242 (2001).
9.
Li Y., Suna Y., Dua F., Yuanc K., Li C., Pulse gradient, large-volume injection, high throughput ultra-performance liquid chromatographic/tandem mass
spectrometry bioanalysis for measurement of plasma amrubicin and its metabolite amrubicinol, J. Chromatogr. A, 1193, 109-116 (2008).
10.
Lovin I., Albu F., Tache F., David V., Medvedovici A. Solvent and salting effects on sample preparation for the determination of fenofibric acid in human
plasma by HPLC-DAD, Microchem. J. 75, 179-187 (2003).
11.
Loeser E, Drumm P. Using strong injection solvents with 100% aqueous mobile phase in RP-LC, J. Sep. Sci. 29(18), 2847-2852 (2006).
12.
Medvedovici A., David Vasile, David Victor, Georgita C. Retention phenomena induced by large volume injection of solvents non-miscible with the mobile
phase in reversed phase liquid chromatography, J. Liq. Chromatogr. Relat. Technol. 30, 199-213 (2007).
13.
Gritti F., Piatkowski W., Guiochon G. Study of the mass transfer kinetics in a monolithic column, J. Chromatogr. A, 983, 51-71 (2003).
14.
Miyabe K., Guiochon G. New model of surface diffusion in reversed phase liquid chromatography, J. Chromatogr. A, 961, 23 – 33 (2002).
15.
Ahmad T., Gritti F., Lin B., Guiochon G., Single component shock layer analysis in elution chromatography, Anal. Chem., 76, 977 – 984 (2004).
16.
David V., Barcutean C., Georgita C., Medvedovici A. Non-miscible solvent large volume injection – HPLC/DAD method for determination of butylated
hydroxyanisole in lovastatin and simvastatin pharmaceutical formulations, Rev. Roum. Chim., 5, 445-451 (2006) .
17.
Udrescu S., Medvedovici A., David V., Effect of large volume injection of hydrophobic solvents on the retention of less hydrophobic pharmaceutical solutes
in RP-LC, J. Sep. Sci., 31, 2939-2945 (2008).
18.
Udrescu S., Sora I.D., David V., Medvedovici A., Large volume injection of hexane solutions in RPLC/UV to enhance on sensitivity of the assay of ginkgolic
acids in Ginkgo Biloba standardized extracts, J. Liq. Chromatogr. Relat. Technol., 33,133-149 (2010).
19.
Udrescu S., Sora I.D., Albu F., David V., Medvedovici A. Large volume injection of 1-octanol as sample diluent in reversed phase liquid chromatography:
Application in bioanalysis for assaying of indapamide in whole blood, J. Pharm. Biomed. Anal., 54(5), 1163–1172 (2011).
20.
Loeser E., Babiak S., Drumm P., Water-immiscible solvents as diluents in reversed-phase liquid chromatography, J. Chromatogr. A, 1216, 3409-3412
(2009).
21.
Ruta J., Rudaz S., McCalley D.V., Veuthey J.L., Guillaume D. A systemathic investigation of the effect of sample diluent on peak shape in hydrophilic
interaction liquid chromatography, J. Chromatogr. A, 1217, 8230-8240 (2010).